Experimental Methods
We have generated a large corpus of scRNA-seq data using our Multiplexed PBMC scRNA-seq Pipeline, based on the 10x Genomics 3' scRNA-seq v3.1 platform, along with antibody-based Cell Hashing reagents from BioLegend for sample multiplexing. Our implementation of this approach is described in (Genge, et al., 2021), based on the approach originally described in (Stoeckius, et al., 2018). This pipeline enables simultaneous processing of up to 24 PBMC samples in a single "Batch," in which up to 13 samples are mixed in each of two "Pools" of cells.
Each Pool includes a Batch Control sample, which is an aliquot from the same single blood draw used across all pipeline experiments, and enables monitoring of batch effects in the resulting data. These pools are then distributed across three 10x Genomics 3' scRNA-seq "Chips", each containing 8 "Wells".
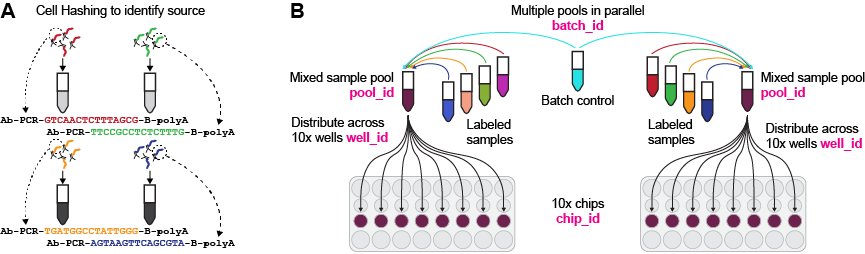 Sample pooling approach and identifiers. A. each sample is labeled with a different cell hashing antibody to enable later identification of sources. B. Each batch may consist of multiple pools of labeled samples, which are then distributed across one or more chips and many wells. Labels in magenta correspond to the batch-related metadata that can be found in our datasets. Figure adapted from (Swanson, et al., 2022).
Sample pooling approach and identifiers. A. each sample is labeled with a different cell hashing antibody to enable later identification of sources. B. Each batch may consist of multiple pools of labeled samples, which are then distributed across one or more chips and many wells. Labels in magenta correspond to the batch-related metadata that can be found in our datasets. Figure adapted from (Swanson, et al., 2022).
From each Well, we generate two sequencing libraries - one for Gene Expression, and the other for Hash Tag Oligonucleotides (HTO). These libraries have common Cell Barcodes that originate from the 10x Genomics GEMs encapsulated in droplets along with PBMC cells. These sequencing libraries are later processed using a pipeline that deconvolutes the original samples. See the Data processing pipeline section for more details.
In many cases, both scRNA-seq and Flow Cytometry are performed using the same aliquots of cells, which are divided and processed on the same day. In these cases, both scRNA-seq and Flow Cytometry data are given the same Batch ID. A detailed protocol describing this joint scRNA-seq and Flow Cytometry Pipeline approach is provided in the article "Optimized workflow for human PBMC multiomic immunosurveillance studies" (Genge, et al., 2021).
Data Processing Pipeline
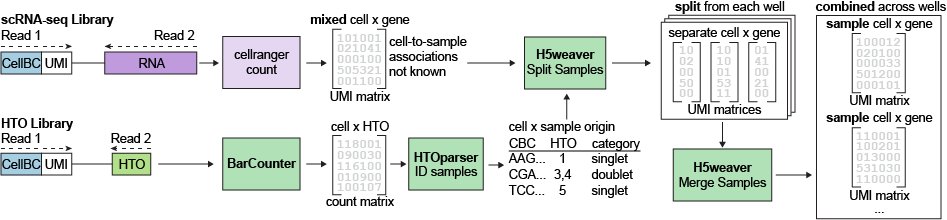
After generating sequencing libraries, we sequenced both the Gene Expression and HTO libraries on Illumina sequencing platforms. Multiplexed sequencing data were prepared using a preprocessing pipeline based on 10x Genomics CellRanger software (version 3.1.0; Released July 24, 2019), with the 10x Genomics GRCh38/hg38 Human Transcriptome Reference (refdata-cellranger-GRCh38-3.0.0; based on the GRCh38 genome reference and Ensembl v93 transcriptome annotations; Released Nov. 19, 2018) to generate cell x UMI count matrices for each Well. To identify cells originating from each sample, we utilized software tools for HTO quantification (BarCounter) and sample demultiplexing (HTOparser, H5weaver) to generate separate HDF5-formatted files for each original sample. This process also removes Cell Barcodes that contain HTOs from multiple samples (doublets or multiplets) or with no detectable sample label (no hash).
 As part of the sample processing pipeline, QC reports are generated to allow review of the quality of data in each 10x Genomics well within each batch. QC reports are reviewed and wells that are low quality are removed prior to final data assembly and labeling.
As part of the sample processing pipeline, QC reports are generated to allow review of the quality of data in each 10x Genomics well within each batch. QC reports are reviewed and wells that are low quality are removed prior to final data assembly and labeling.
Detailed descriptions of this demultiplexing process are provided in the article "BarWare: efficient software tools for barcoded single-cell genomics" (Swanson, et al., 2022)
The full implementation of the preprocessing pipeline utilized in the Human Immune System Explorer is available in Github, and utilizes the following components:
Pipeline wrapper scripts
BarWare Pipeline: For use outside of the HISE environment, we recommend using the BarWare pipeline repository, which encapsulates specific components of the cell hashing pipeline to simplify sample demultiplexing:
AllenInstitute/BarWare-pipeline
10x scRNA-seq Pipeline wrappers: Scripts used in HISE pipelines for processing 10x Genomics scRNA-seq data produced by Cell Ranger
aifimmunology/tenx-rnaseq-pipeline
Cell Hashing Pipeline wrappers: Scripts used in HISE pipelines to analyze cell hashing oligos and for splitting and merging scRNA-seq data based on sample assignments
aifimmunology/cell-hashing-pipeline
Applications
BarCounter: Used to efficiently compute counts for Hash Tag Oligo (HTO) libraries
AllenInstitute/BarCounter-release
R Packages
batchreporter: Used to generate detailed batch QC reports for review and approval of datasets
aifimmunology/batchreporter
H5weaver: Used to add metadata and split and reassemble count data and metadata based on sample identity.
aifimmunology/H5weaver
HTOparser: Used to identify samples based on Hash Tag Oligo (HTO) counts.
aifimmunology/HTOparser
SeuratLabeler: A wrapper around Seurat v4 and a reference dataset used to add initial cell type labels
aifimmunology/SeuratLabeler
References
Genge PC, Roll CR, Heubeck AT, Swanson E, Kondza N, Lord C, et al. Optimized workflow for human PBMC multiomic immunosurveillance studies. STAR Protoc. 2021;2: 100900.
doi:10.1016/j.xpro.2021.100900
Stoeckius M, Zheng S, Houck-Loomis B, Hao S, Yeung BZ, Mauck WM 3rd, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018;19: 224.
doi:10.1186/s13059-018-1603-1
Swanson E, Reading J, Graybuck LT, Skene PJ. BarWare: efficient software tools for barcoded single-cell genomics. BMC Bioinformatics. 2022;23: 106.
doi:10.1186/s12859-022-04620-2